ADN et archéologie
L’apport de la science génétique à l’archéologie
par J.-Y. Moisan
|
L’archéologie est une science qui interroge beaucoup les autres sciences pour savoir ce qu’elles pourraient lui apporter. Ainsi les sciences physiques ont permis la datation des fossiles (14C, thermoluminescence, …). Il n’est donc pas surprenant que très tôt les archéologues se soient intéressés aux récentes découvertes biologiques sur le génome humain. Deux questions en particulier ont été formulées : les différences observées dans le génome suivant les diverses populations dans le monde sont-elles significatives de leurs origines ? Est-il possible d’analyser des fossiles et d’en tirer des informations sur l’évolution de l’espèce humaine ? C’est ce que l’on va tenter de discuter. Préambule - Rappels sur ce qu’est le génome Un organisme vivant est composé de cellules. Les premiers êtres monocellulaires sont apparus il a 3,5 milliards d’années : aujourd’hui les plantes et animaux les plus évolués sont composés de milliards de cellules (environ 1014, soit 100.000 milliards, chez l’homme), ayant des fonctions différenciées dans l’organisme. Très schématiquement une cellule animale est composée d’un noyau (où est rassemblée la majorité de l’information transmissible), entouré d’un cytoplasme (dans lequel on trouve différents organites, dont les mitochondries : nous en reparlerons), le tout enfermé dans une membrane. Intéressons nous d’abord au noyau : il contient tous les chromosomes, porteurs de l’essentiel de l’information cellulaire. Chez l’homme, les chromosomes sont au nombre de 46 et mis bout à bout, ces filaments font 35 cm de longueur. Chaque chromosome est constitué d’une molécule d’ADN linéaire. L’ensemble des chromosomes d’une cellule s’appelle le génome, propre à chaque personne. On notera immédiatement que la paire de chromosome 23 est spécifique du sexe de la personne : chez la femelle la paire est composée de 2 chromosomes X ; chez le mâle elle comprend un chromosome X et un chromosome Y. Lors de la reproduction, si le père donne un chromosome X, l’enfant sera une fille ; s’il donne un chromosome Y, l’enfant sera un garçon. On voit donc que le chromosome Y est donc spécifique de la lignée paternelle et sera le même d’une génération à l’autre dans la filiation paternelle. Dans le cytoplasme de la cellule, les mitochondries seraient, à l’origine, des bactéries que les cellules primitives auraient phagocytées. Leur rôle est important dans le métabolisme de la cellule. Mais surtout elles possèdent leur propre ADN, l’ADN mitochondrial ou ADN-mt. Or, contrairement au chromosome Y, l’ADN-mt est reçu de la mère lors de la reproduction. Il sera donc caractéristique de la lignée maternelle. L’ADN-mt est constitué des 16.569 paires de base (comparé à plus de 3 milliards pour l’ADN nucléaire). Mais, la plupart des cellules contient plus de quelques centaines de mitochondries qui chacune contient 8 chromosomes semblables. Lorsque l’ADN est dégradé, ce sera un matériau de choix. Allons un peu plus loin dans la description de l’ADN (voir 2), jusqu’à l’échelle atomique ! Cette molécule est formée de deux chaînes enroulées l’une sur l’autre; elle ressemble à une échelle où les barreaux sont la combinaison de deux bases associées. L’ADN stocke l’information génétique à l’aide d’un alphabet de 4 lettres (A : Adénine ; T : Thymine ; C : Cytosine et G : Guanine) formant des mots de 2 lettres : les nucléotides associés par deux. La figure 2 montre une série de 4 associations de bases : la répétition d’une telle ‘série’ va constituer un segment. Un tel segment spécifique et localisé en un endroit bien défini de la molécule est appelé ‘gène’. Un gène est donc une unité définie, localisé en un point précis d’un chromosome, auquel est lié un caractère précis de l’individu et transmissible par voie héréditaire. Les gènes sont portés sur des parties dites ‘codantes’ de l’ADN. Des parties non-codantes occupent une partie des chromosomes, dont l’altération ne semble pas avoir de conséquences médicales. Toutefois, sur ces dernières parties on peut aussi identifier des séquences bien définies qui peuvent être caractéristiques d’un ensemble d’individus. Finalement, nous aurons à notre disposition (pour ce qui nous intéresse) deux sources d’information’ : celle de l’ADN-Y, hérité de notre père et celle de l’ADN-mt, toujours hérité de notre mère. Après la formation de la cellule mère de chaque individu, la croissance de l’embryon, puis de l’individu sera assurée par division cellulaire et chaque cellule contient le même génome quelle que soit sa fonction finale dans l’organisme (à l’exclusion des cellules dites ‘gamètes’, propres à la reproduction). N’importe quelle cellule d’un organisme permettra donc de savoir beaucoup sur ses origines. Mutations génétiques et évolution Nous connaissons tous des exemples de maladies génétiques. Ainsi la trisomie 21 : trois chromosomes s’associent au lieu de former une paire lors de la fusion des gamètes (ce sont les cellules reproductrices, venant du père - le spermatozoïde – et venant de la mère – l’ovule -). D’autres ‘accidents’, qui ne modifient que légèrement un gène, sont lourds de conséquences sur le plan médical et très invalidants pour le porteur de ces anomalies génétiques. Toutes ces anomalies ont peu de chance d’être transmises aux générations suivantes, pour des raisons que l’on comprend aisément. Dans de tels cas, la copie n’est pas conforme à l’un des deux originaux. Heureusement, les conséquences ne sont pas toujours aussi catastrophiques. Ces modifications, qu’on appelle des ‘’mutations’’, peuvent n’avoir aucun effet d’un point de vue médical ; elles peuvent aussi apporter un plus, en provoquant chez le porteur, par exemple, une meilleure adaptation à l’environnement. Ces modifications n’ont pas toutes la même importance : - ce peut être une simple modification d’une paire de nucléotide ; - ce peut être une inversion de grandes séquences d’ADN ; - ce peut même être une fusion ou une coupure d’un chromosome (nous en verrons un exemple). Ces mutations n’affectent pas uniquement les chromosomes du noyau cellulaire. Elles affectent (et même plus souvent) l’ADN-mitochondrial. Et une mutation qui sera transmise aux générations suivantes peut devenir fréquente au sein d’une population isolée et faible en nombre. Ce sera moins probable dans une population nombreuse. Un ensemble de mutations observées sur un endroit précis du génome pourra servir de ‘marqueur’ et être spécifique d’une population. Un ensemble de marqueurs pourra définir un haplotype : les porteurs de cet haplotype formerons un haplogroupe Nous reviendrons sur cette notion d’haplogroupe. Ces mutations se produisent ‘au hasard’ : ce sont des phénomènes relevant des sciences statistiques. Sur un nombre élevés de générations la probabilité que se produisent des mutations est maintenant établie : le rythme en est connu. Donc les ‘écarts’ (les mutations) entre deux caryotypes pour des individus (ayant le même ancêtre) seront plus ou moins nombreux suivant que le nombre de générations qui séparent ces deux individus de leur ancêtre commun est plus ou moins grand. On dispose ainsi d’une horloge : avec une estimation de la durée intergénérationnelle, le nombre de différences, sur l’ADN-mt ou l’ADN nucléaire, signifiera le temps qui séparent deux populations d’aujourd’hui de la population à l’origine de celles-ci. Nous verrons des exemples de l’utilité de cette horloge par la suite. Variabilité D’abord une évidence ; tous les hommes sont génétiquement apparentés : ils peuvent se marier entre eux et avoir des enfants. Mais il est aussi évident que vous verrez aisément des différences entre des personnes originaires de continents différents, mais plus difficilement s’ils viennent de pays voisins. D’ailleurs ces différences visibles furent à l’origine de la notion de races. Prenons maintenant la taille de l’individu comme paramètre variable. Les Pygmées sont petits. Mais un grand Pygmée peut être plus grand qu’un petit dans une population voisine. D’autre part, l’écart en taille dans la population Pygmée (la variabilité en taille) est plus faible chez les Pygmées que dans la population voisine. Notre code génétique propre est commun à l’ensemble de l’humanité à 99,99 %. Les variations individuelles ne portent que sur 0,01 % du génome humain ! Différencier des populations entre elles sur la base de leur ADN, exigera donc une analyse précise avec des outils performants ! Enfin, à cause des mutations survenant périodiquement, avec le temps, à l’intérieur d’une population, l’évolution du patrimoine génétique commun évolue et les écarts entre les individus augmente : la variabilité augmente. Ainsi une population ‘’jeune’’ (issue depuis peu d’un groupe originel restreint) aura une variabilité plus faible qu’une population ‘’ancienne’’, c’est-à-dire qui vit et se reproduit sur un territoire clos depuis longtemps (en nombre de générations) tout en étant issue de la même manière d’un groupe originel restreint. Fragilité de l’ADN S’il est une molécule ‘vivante’, c’est bien l’ADN, mitochondrial et nucléaire. Pour cette raison, comme toute vie, l’ADN est fragile : elle évolue dans un milieu où les réactions biochimiques sont constantes … et pas seulement lorsque l’organisme est vivant, mais après l’arrêt des fonctions vitales de l’organisme. L’ADN est chimiquement instable. Par exemple, les coupures de chaînes sont fréquentes. Mais la défense de l’organisme existe : des enzymes se chargent des réparations, tant que l’organisme est vivant. Un exemple : le nucléotide C (la cytosine) peut se transformer en U (l’uracile) qui ne participe pas à la molécule d’ADN. Des enzymes se chargent de la réparation et l’uracile est éliminé de la cellule. On va la retrouver dans les déchets de l’organisme, c’est-à-dire dans l’urine. Une analyse permet alors de chiffrer le nombre ‘d’attaques’ de cette espèce subie : c’est environ 10 000 par jour pour un individu. Chiffre à rapprocher toutefois du nombre de nucléotides par cellule et du nombre de cellules dans notre corps. Mais au décès de l’individu, les mécanismes de réparation vont s’arrêter : en quelques jours l’ADN va être fracturé en morceaux de plus en plus petits. Parallèlement, les défenses contre les bactéries ne fonctionnent plus et ces dernières dégradent toute la matière organique. Il faudra donc des conditions bien particulières (et beaucoup de chance) pour que l’on puisse retrouver de l’ADN ‘analysable’ dans un fossile. On sait par exemple, qu’un milieu humide et acide est très défavorable à la conservation de l’ADN : c’est le cas des hommes trouvés dans les tourbières de l’Europe du Nord. Par contre la survie est plus longue dans un milieu sec et basique : ce peut être le cas d’une grotte calcaire. Enfin, la congélation, dans le permafrost par exemple, est favorable aussi à la conservation. Évolution de la population mondiale Enfin, peut-être de façon étonnante, en tout cas a priori bien éloignée de la génétique, nous discuterons de l’évolution de la population mondiale. Nous sommes aujourd’hui 7 milliards d’humain sur la planète. D’après le tableau de la figure 3, nous n’étions qu’environ 1 milliard en 1800 et moins de 100 millions au temps du Christ. Le schéma de la figure 3 n’est donc pas très lisible. Sur la figure 4 (obtenue auprès de l’INED, Institut National des Etudes Démographiques), le schéma est plus lisible dans la mesure où l’ordonnée est en coordonnées logarithmiques, c’est–à-dire que les unités d’échelle sont des multiples de 10 (à chaque unité, le nombre est multiplié par 10). On y voit alors très clairement qu’avec l’arrivée de la révolution néolithique (il y a environ 10.000 ans), la population va passer de 7 millions d’individus aux 7 milliards d’aujourd’hui (soit 1.000 fois plus en 10.000 ans). Pendant les 25.000 ans précédents (soit environ 1.000 générations !), elle était pratiquement stable, si nous oublions les évènements tels qu’épidémies, alea climatiques …Et pendant la période de -65.000 à -40.000 ans, c’est environ 600.000 humains qui peuplent la planète, soit un nombre 10.000 fois plus faible qu’aujourd’hui. Quel intérêt de tels chiffres ? Prenons un exemple, traité de façon grossière mais qui permet de montrer certaines réalités. La France d’aujourd’hui n’était peuplée que d’environ 65.000 personnes au Paléolithique, et d’environ 6.500 individus entre -40.000 et -65.000 ans. En faisant l’hypothèse (pour simplifier) que les familles (ou clans ou tribus, …) formaient des groupes de 65 individus, il n’y avait sur le territoire français, qu’environ 100 groupes. Ce calcul semble bien grossier. Et pourtant lorsque les colons danois découvrirent le Groenland au XVIe siècle, ils trouvèrent sur la côte ouest 499 Inuits. Or cette côte est aussi longue que celle qui va de la Bretagne au Portugal. Rapportée à la surface de la France actuelle, la même densité de population y ferait vivre un peu moins de 7.000 personnes ! Or – nous le verrons – c’est en ces périodes que l’être humain va se répandre sur la terre. La notion de population dispersée, où l’évolution biologique pouvait affecter des groupes de façon différente pour les uns et pour les autres, prend tout son sens. Ces préambules permettent d’aborder maintenant deux questions : 1. Que peut apporter la connaissance de l’ADN des hommes actuels à la connaissance de notre histoire. 2. Que pourrait apporter la connaissance de l’ADN ancien (celui des fossiles) à l’histoire de l’humanité et –pourquoi pas- préciser notre origine. Rappelons-nous le scandale provoqué par la publication par Charles Darwin, qui avançait que l’Homme descendait du singe et venait d’Afrique ! En fait, l’homme ne ‘descend’ pas du singe, mais il partage avec les grands singes un ancêtre commun. C’est ce que l’archéologie a montré depuis plusieurs dizaines d’années ! Depuis l’ancêtre commun, l’homme a bien sûr évolué comme les grands singes de leur côté. Si Darwin avait disposé d’analyses de l’ADN de l’homme et du chimpanzé actuels, alors une réalité bien pire aurait effrayé nos ancêtres. En effet, on peut aligner côte à côte 95 % des séquences des ADN respectifs de l’Homme et du chimpanzé et les différences ne portent que sur 1,2 % des gênes. Notre humanité ne porte donc que sur 1,2 % de nos gênes ! Mais ces différences portent sur 3 milliards de nucléotides, ce qui laisse quand même beaucoup de gênes différents ! 2 – L’origine de l’homme Que peut nous dire l’ADN des hommes vivants aujourd’hui Depuis qu’il est possible (à assez peu de frais !) de faire le génome des hommes d’aujourd’hui, un programme international a été lancé (le Projet Génome) pour analyser le génome de personnes réparties sur la planète et appartenant aux diverses ethnies existantes. Le nombre de ces analyses se chiffre évidemment en milliers. Parallèlement le génome de diverses espèces animales est étudié, et bien sûr celui des proches de l’homme, les grands singes. Leur génome est le plus proche du nôtre parmi tous les autres mammifères et exprime notre parenté. Intéressons-nous à l’écart entre homme et chimpanzé. A partir des horloges génomiques dont nous avons parlé ci-dessus, l’ancêtre commun entre chimpanzés et nous aurait vécu il y a 4 à 7 millions d’années. Depuis cette date, le génome du chimpanzé –au fil du temps et des mutations de l’ADN- a évolué dans la population restée confinée en petits groupes dans les forêts africaines. Si maintenant on ajoute l’ADN des gorilles et des orangs-outangs, les écarts observés conduisent à des ancêtres communs avec les précédents (hommes et chimpanzés) vivants il y a 7 à 8 millions d’années pour les premiers et 12 à 14 millions pour les seconds. Ceci consolide l’arbre phylogénétique, représenté sur la figure 5. Ceci ne contredit pas ce que l’archéologie nous a appris à partir de l’étude des fossiles. La génomique vient ici consolider ce résultat. Mais nous avons laissé au passage une question qui concerne la variabilité au sein de l’espèce humaine. Mais un résultat concernant la variabilité au sein de chaque espèce pose problème. Au laboratoire de Génétique de l’Institut Max-Plank d’anthropologie évolutionniste de Leipzig, en Allemagne, une étude a porté sur le chromosome X (1 seul chez l’homme : ceci réduit le domaine de l’analyse), de 69 hommes choisis en fonction de critères linguistiques. Entre deux individus, l’écart moyen est de 3,7 différences sur ce chromosome. On remarque aussi que si l’analyse se concentre sur les africains, alors l’écart est sensiblement plus grand que chez les non-africains. Nous y reviendrons. Le même travail chez 30 chimpanzés donne un autre résultat : l’écart moyen entre deux individus est de 13,4 différences. Or les chimpanzés sont environ 200 000, vivant uniquement dans les forêts équatoriales d’Afrique. Par contre, nous, les humains, sommes 7 milliards, répartis sur toute la surface du globe. Même si le nombre des échantillons est petit par rapport aux populations visées, le résultat est significatif. La géographie aurait pu nous faire attendre un résultat contraire : une variabilité plus grande chez l’homme que chez le chimpanzé. Comment expliquer ce résultat sur le critère de la variabilité, qui apparaît donc nettement plus grande chez le chimpanzé, (moins nombreux et géographiquement localisés) que chez les hommes (beaucoup plus nombreux et universellement répartis) ? Ceci voudrait dire que les chimpanzés ont un ancêtre commun relativement plus éloigné dans le temps que les hommes. Or nous avons pourtant un ancêtre commun, les chimpanzés et nous ! Il nous faudra revenir sur ce résultat. Le peuplement de la terre par l’espèce humaine. Il ne faut pas rêver : la fragilité chimique de l’ADN est telle que nous ne verrons sans doute jamais le génome de Lucy. Cette gentille australopithèque (à la démarche sans doute assez peu élégante) vivait il y a 3,2 millions d’années, dans un climat chaud, exposée aux pluies et donc peu propice à la conservation de son ADN ! Il semble (nous n’avons pas de fossiles hors d’Afrique) qu’elle et ses compagnons ne sont pas sortis du continent africain. L’archéologie nous apprend aussi qu’est apparu, issu des australopithèques, l’homo erectus, qui lui a colonisé une grande partie des continents. Cet ancêtre a quitté l’Afrique de l’Est il y a un peu moins de 2 millions d’années. Il est arrivé en Europe (d’après les plus anciens fossiles) un peu avant 1 millions d’années. Mais il était déjà en Asie du Sud-est autour de 1,5 millions d’années. (voir figure 6) Il y a sans doute peu de fossiles. Mais rappelons-nous que la population devait aussi être très peu nombreuse. De plus les conditions pour qu’un fossile puisse se conserver sur d’aussi longues durées sont drastiques. Et ne parlons pas de l’ADN ! Ce que nous apprend aussi, l’archéologie, c’est que ces populations d’homo erectus ont évolué pendant ces 1 à 1,5 millions d’années et par exemple en Europe va lui succéder quelqu’un dont on va beaucoup parler, l’homme de Neandertal. Sur les autres continents serait apparus des cousins de l’homme de Neandertal. C’est ce qu’on appelle l’hypothèse (ou théorie) multirégionaliste. : Homo Erectus évolue localement vers des formes plus évoluées, localement en Europe vers l’homme de Neandertal, celui-ci évoluant ensuite vers homo sapiens. Déjà avec les connaissances de l’archéologie, cette hypothèse n’explique pas ce qu’on observe, au moins en Europe, à savoir une disparition de l’homme de Neandertal remplacé par homo sapiens sapiens. (Voir figure 7, hypothèse B) Une autre hypothèse est celle de l’origine unique (figure 7, hypothèse A). . Homo erectus évolue localement vers des formes d’homo sapiens archaïques, dont celle de Neandertal en Europe. Mais il disparaît au profit d’un ‘cousin’, venu ‘d’ailleurs’ et qui va peupler tous les continents. C’est l’hypothèse dite aussi ‘Out of Africa’, en supposant que cette population est originaire d’Afrique. La troisième hypothèse, dite réticulée, est un peu un mélange des deux précédentes. Elle suppose des métissages des populations, à l’exclusion de celles d’Europe, excluant donc de fait l’homme de Neandertal. Comment trancher ? Ce que nous avons dit ci-dessus à propos de la variabilité dans le génome humain peut-il apporter quelques lumières ? Le faible écart observé dans le génome humain conduit à estimer que l’ancêtre commun aux 7 milliards d’homme sur la terre aujourd’hui vivait il y a environ 200 000 ans. De plus, la variabilité dans le génome humain apparaît un peu plus élevée sur le continent africain que sur le reste de la planète. L’hypothèse multirégionale supposerait un ancêtre commun qui serait homo erectus, vivant il y a plus d’un million d’années. Or ce n’est pas ce qu’on observe. Nous avons donc une origine commune unique, apparue bien après homo erectus. Si l’on considère l’hypothèse ‘Out of Africa’, on peut alors expliquer la variabilité légèrement plus grande en Afrique qu’ailleurs : homo sapiens sapiens évolue à partir d’homo erectus, à travers de multiples métissages, jusqu’au moment, il y a environ 200 000 ans où il ‘décide de partir peupler les autres continents’ et se substituer aux autres populations issues, localement d’homo erectus. L’homme moderne (homo sapiens sapiens) est apparu en Afrique, il y a un peu plus de 200 000 ans et en est sorti après il y a environ 150 000 à 200 000 ans. Nous sommes donc bien, tous issus d’une population (faible) venant d’Afrique et qui a colonisé tous les continents. Les tenants de la théorie multirégionale ont eu parfois quelques difficultés à accepter le verdict ! Le déferlement de l’homo sapiens sapiens. Ensuite, comment cette vague a-t-elle déferlé sur les continents ? Peut-on en savoir plus sur les itinéraires choisis par l’homme moderne pour peupler la terre ? Le ‘projet génome’ (dont nous avons parlé ci-dessus) a permis de mettre à disposition des chercheurs des milliers de génomes humains, venant de personnes choisies au milieu de populations différentes par leurs ethnies, leur langues, … On ne sera pas surpris d’apprendre qu’il est alors possible de caractériser des groupes humains à partir d’une collection de gênes qui leurs sont particulièrs : c’est ce qu’on appelle des ‘haplogroupes’. Les groupes humains, localisés géographiquement, et faibles en nombre, ont évolué, grâce aux mutations de leur ADN : ainsi lors d’un déplacement vers une nouvelle contrée, une population d’individus va évoluer différemment de la population dont elle est originaire et former un nouvel haplogroupe, issu du précédent. Le tableau ci-contre explique les résultats obtenus. Les ‘SNP’ du tableau 8 sont des ‘marqueurs’, c’est-à-dire des groupes de gênes spécifiques d’un haplogroupe. Il faut bien noter que cette étude porte uniquement sur le chromosome Y, le chromosome ‘masculin’. Sur le tableau 8, les haplogroupes sont classés suivant leur occurrence géographique et sur la figure 9, l’arbre phylogénétique de ces haplogroupes est reconstruit. Par exemple les groupes A et B, les plus anciens sont trouvés en Afrique. Le groupe I (M170), plus récent est trouvé en Europe, ainsi que le groupe O (M175) en Asie. Le groupe Q (M242), le plus récent sur l’arbre phylogénétique, est spécifique de l’Amérique. La sortie d’Afrique apparaît ainsi clairement. De même, la variabilité du génome s’est construite lors de la diffusion de l’homme moderne à travers les continents. On peut donc de cette manière décrire l’itinéraire des hommes modernes dans leur colonisation des territoires. La figure suivante en est un exemple : avec un départ d’Afrique de l’Est vers - 60 000 ans, l’homme moderne arrive en Europe aux alentours de - 20 000 ans. Les différents marqueurs, notés sur la carte, permettent de suivre son parcours. Notons bien que ces marqueurs sont ceux que l’on trouve dans les populations actuelles. Quelques remarques :
Après l’exemple européen, prenons-en un autre qui pourrait apparaitre simple, celui de la colonisation de l’Amérique (celle des Amérindiens et non des Espagnols … !). L’homme est présent 20.000 ans avant notre ère dans l’est de la Sibérie. Les glaciations font plutôt le passage facile vers l’Amérique et donc un schéma de diffusion comme sur la figure 11 semble évident. Or déjà les nombreuses langues parlées appartiennent à 3 groupes différents : esquimau-aléoute (nord), na-déné (Apaches et Navajos, à l’ouest) et amérindes (centre et sud). Ces groupes se distinguent aussi par des morphologies différentes. Ceci peut s’expliquer par des vagues successives de populations, suivant des routes différentes : le long des côtes ou par l’intérieur du continent. Mais l’étude par les variations de l4ADN est très délicate, du fait entre autres de faibles variations attendues sur une période aussi courte. Mais la colonisation de la planète s’est poursuivie jusqu’à des époques ‘récentes’ puisque après l’Amérique il y a seulement environ 20 000 ans, l’Océanie n’a été peuplée qu’il y a environ 5 000 ans. Et enfin les hommes sont arrivés sur l’Ile de Pâques vers 1 200 de notre ère ! Et pour préciser touts ces routes de colonisation, bien du travail reste çà faire. 3 – L’origine de l’homme Que peut nous dire l’ADN ancien ? Il faut comprendre : est-il possible de récupérer sur des fossiles de l’ADN et d’en apprendre quelque chose de nos origines ? D’emblée, trois problèmes de taille seront à résoudre :
Ces progrès des outils d’analyse ont confortablement aidé les généticiens et compensé partiellement les difficultés liées à la qualité et la quantité des échantillons. Mais ne lèvent pas le problème de la contamination. A la poursuite du génome de l’homme de Neandertal. Bien des tentatives étaient programmées dans divers laboratoires : il faut dire que celui qui publierait le génome de Neandertal allait acquérir immédiatement une notoriété internationale ! Le laboratoire qui y est parvenu est celui de Svante Pääbo, un norvégien, généticien évolutionniste, directeur du département de Génétique de l’institut Max-Plank d’anthropologie évolutionniste de Leipzig. Il s’était auparavant essayé sur des momies égyptiennes, sur Ötzi, vieux de 5 300 ans mais conservé dans les glaces des Alpes. Ötzi avait été trouvé en septembre 1991 par des randonneurs. Le fossile était conservé dans le froid à Innsbruck et S Pääbo avait été autorisé à faire quelques prélèvements aux fins d’analyse de l’ADN de cet homme. L’étude fut très laborieuse. Les échantillons superficiels étaient beaucoup trop contaminés pour être significatifs. Des échantillons internes ont toutefois permis d’obtenir une séquence de 300 nucléotides de l’ADN-mt de Ötzi. Un autre laboratoire – à la demande de S Pääbo - a validé ce résultat. Comparé aux 16 500 nucléotides d’un ADN-mt et pour un fossile relativement ‘jeune’, le bilan peut paraître un peu court, pour constater très peu d’écart avec un européen actuel. Mais il faut aussi remarquer que 5 300 ans, dans l’évolution, est un temps court : c’est environ 250 générations et donc le nombre de mutations attendues et observables est tel que le génome de Ötzi est certainement très proche des européens d’aujourd’hui. Difficile dans ces conditions de savoir, à cause des contaminations toujours possibles, ce qui lui est propre. Mais l’équipe de chercheurs, dans ce travail de 1993, a acquis expérience et méthodes, celles-ci devenant de plus en plus fiables. Dans le même temps les outils à leur disposition devenait de plus en plus performants. Ainsi, les musées devenaient-ils de véritables banques de gênes. En 1996, S Pääbo obtient la possibilité de prélever une partie de l’humérus droit de l’homme de Neandertal, le fossile type découvert en 1853, un échantillon de 3,5 grammes. A la fin de l’année, une séquence d’ADN-mt obtenue est originale : elle ne ressemble pas à l’ADN-mt de l’homme actuel. C’est une séquence de 379 nucléotides (à comparer aux 16 500 nucléotides d’un ADN-mt !) : mais cette séquence peut parfaitement être comparée à la séquence équivalente (en position dans l’ADN) d’humains modernes. Ce qui est fait bien sûr, avec les mêmes séquences de 2051 génomes d’hommes actuels. Les résultats sont les suivants :
Ce résultat fait l’objet d’une publication en juillet 1997. Il est confirmé par une autre publication en mars 2000, par un laboratoire anglais. Ce qui est important, c’est qu’il est ainsi démontré la possibilité d’étudier l’ADN de fossiles ayant vécu il y a environ 30 000 ans. Certes il restait, en 2000, beaucoup à faire pour connaître le génome complet de l’homme de Neandertal. Mais ce premier résultat était important. Svante Pääbo obtient, en 1999, onze échantillons d’os de Néandertaliens, venant d’une grotte en Croatie. Les ossements sont datés d’environ 30 000 à 40 000 ans (ce qui est un peu jeune pour des hommes de Neandertal). Mais ils provenaient d’une grotte calcaire, donc un milieu basique plus favorable à la conservation de l’ADN. De plus, les ossements portaient des traces caractéristiques de rites anthropophagiques : ceci signifie que les ossements ont été entreposés sans chair dans la grotte et donc la contamination par des bactéries était de ce fait limitée. Une nouvelle partie de l’ADN-mt est alors obtenue. Donc finalement, à partir des ossements de trois hommes de Neandertal, trois séquences de l’ADN-mt sont disponibles à la fin de l’année 2000. En étudiant ces trois séquences, on constate : Un écart de 3,7 % de nucléotides différents entre les 3 Néandertaliens ; Un écart moyen de 3,4 % de nucléotides différents chez l’homme moderne en les sélectionnant trois par trois ; Un écart de 14,8 % chez le chimpanzé ; Un écart de 18,6 % chez le gorille. Il est donc clair que la variabilité chez l’homme de Neandertal et chez l’homme moderne est sensiblement la même, signifiant qu’ils partagent le même ancêtre commun, plus récent que celui des chimpanzés et encore plus récent que celui des gorilles. Ceci est tout à fait en accord avec l’arbre phylogénétique présenté sur la figure 5 et obtenu par l’analyse des hommes et singes actuels. L’homme de Neandertal vient s’y insérer comme l’archéologie le fait attendre. Enfin en 2008, Svante Pääbo obtient la première séquence complète de l’ADN des mitochondries de l’homme de Neandertal. Information importante : aucune preuve d’un apport génétique de l’homme de Neandertal à l’homme moderne n’est visible. Ceci confortait Svante Pääbo dans son idée qu’aucun métissage n’avait eu lieu. Mais la réponse définitive ne pouvait venir que par l’analyse de l’ADN nucléaire. Ce n’est plus 16 500 nucléotides qu’il faut aligner mais 3 milliards ! Heureusement, pendant la décennie 2000 – 2010, les progrès des outils d’analyse sont considérables. Parallèlement, les outils informatiques nécessaires progressent de la même façon. En 2006, Svante Pääbo montre alors qu’il est possible de faire ce travail, mais avec une très petite séquence (0,0003 % du génome, mais tout de même 10 000 nucléotides). Et finalement en 2010, Svante Pääbo publie la première description approximative de l’ADN entier du noyau d’une cellule de Neandertal. Cet ADN a été isolé à partir des ossements de trois individus venant de la fameuse grotte de Vindija en Croatie. Or pendant treize ans, Svante Pääbo avait toujours pensé qu’aucune trace de Neandertal n’était présente dans l’ADN de l’homme moderne. Il doit convenir pourtant que de 1 à 3 % de notre ADN d’aujourd’hui nous vient de Neandertal. « Cela contredisait ce que moi-même j’avais cru être vrai. Les hommes et femmes de Neandertal ne s’étaient pas totalement éteints. Leur ADN persistait aujourd’hui dans l’humanité » écrivit Svante Pääbo Encore quatre ans et, au début de 2015, Svante Pääbo publie l’ADN complet d’une femme de Neandertal, vieille de 50 000 ans. Cette fois la précision de l’analyse est tout à fait comparable à celle obtenue avec des échantillons d’hommes d’aujourd’hui. Que peut-on en déduire ?
Toutes ces analyses des ADN anciens et des ADN des hommes d’aujourd’hui suggèrent que les derniers ancêtres communs à tous vivaient il y a environ 600 000 ans et donc probablement en Afrique. Deux groupes se sont formés : l’un a migré vers l’Eurasie et sa lignée donnera naissance à l’homme de Neandertal. L’autre restera en Afrique et évoluera vers l’homme moderne. Cet homme moderne migre alors vers l’Eurasie et rencontre alors son cousin Neandertal. Quand et où a eu lieu le métissage ? Est-il possible d’y répondre ?
Plus tard, une nouvelle vague quitte l’Afrique vers – 60 000 et cette fois rencontre en Galilée l’homme de Neandertal et les deux populations se métissent. La population de Neandertal semble en position de faiblesse (pour des raisons qui vont conduire, à sa disparition) ; ce qui n’est pas le cas de la population de l’homme moderne. C’est en effet à ce moment-là qu’on observe une forte augmentation de la population mondiale (voir figure 4). Cette vague joue comme un rôle de rouleau compresseur, qui va noyer ce qui reste de l’homme de Neandertal.
Bien évidemment ce scenario reste à affiner, mais dans l’immédiat, il rend compte des observations faites dans l’analyse du génome des populations concernées, mais aussi des résultats de fouilles archéologiques dans les territoires en question. Un nouveau venu Dans les monts de l’Altaï, aux confins de la Russie, de la Mongolie, du Kazakhstan et de la Chine, des archéologues russes avaient trouvés des ossements dans des grottes. Le responsable de cette équipe avait remis à Svante Pääbo quelques fragments d’os provenant de ces grottes. L’analyse de l’ADN-mt contenu dans ces fossiles indique qu’ils se trouvent appartenir à un Neandertalien. Ce résultat repousse tout à coup de 2 000 km vers l’Est la zone communément admise alors comme territoire occupé par l’homme de Neandertal. Ceci a fait l’objet d’une publication en 2007. Au printemps de 2009, le même archéologue russe fait parvenir à Svante Pääbo un petit fragment d’os, trouvé dans la grotte de Denisova, voisine de la grotte précédente qui avait été occupée par un homme de Neandertal. C’était un petit morceau de phalange, comme le montre la figure 14. Malgré la petite taille de l’échantillon, une grosse quantité de fragments d’ADN-mt (30 443 au total) a permis de reconstruire l’ADN-mt de cet individu (en fait un enfant). Alors que les chercheurs s’attendaient à trouver à nouveau un Néandertalien, ce qu’ils avaient sous les yeux ne ressemblait ni à l’ADN-mt de l’homme de Neandertal, ni à celui d’un homme moderne. Alors que l’ADN-mt de l’homme de Neandertal présente 202 positions nucléotidiques différentes avec l’homme moderne, cet homme de Denisova en présentait 385, soit presque le double. Cet enfant de Denisova n’était donc ni un Néandertalien, ni un homme moderne. En supposant que les hommes et les chimpanzés ont divergé il y sept millions d’années, le même calcul conduit à une divergence entre Neandertal et l’homme moderne il y a environ un demi-million d’années, mais la divergence entre l’homme de Denisova et l’homme moderne aurait eu lieu il a un million d’années. C’est-à-dire pratiquement au moment où l’homo erectus arrivait en Eurasie. Ce résultat a fait l’objet d’une publication en avril 2010. Mais la preuve évidente que la grotte de Denisova abritait bien un nouvel individu jusqu’alors inconnu ne pouvait être définitive qu’avec la description de son ADN nucléaire. Entre temps, Svante Pääbo reçoit en janvier 2010, une molaire trouvée dans la même grotte neuf ans plus tôt. Cette molaire, aux dires des archéologues ne ressemblait pas à une molaire d’homme de Néandertal, encore moins à celle d’un homme moderne. Avec la recherche de l’ADN nucléaire dans le fragment d’os qui avait servi à la production de l’ADN-mt de l’homme de Denisova, il apparaît d’abord qu’il y avait très peu d’ADN-Y. Ce qui voulait dire que cet os appartenait à une fillette (et que la contamination était très faible !). Il est apparu que le génome de cette fillette était plus proche de celui de l’homme de Neandertal que de celui de l’homme moderne. Ensuite, un autre résultat est surprenant : l’individu de Denisova partageait plus de marqueurs avec un Papou qu’avec un chinois, un européen ou un africain. Pour s’assurer de la validité de cette découverte, le génome de l’homme de Denisova est alors comparé aux génomes de 938 humains appartenant à 53 populations différentes dans le monde. Et effectivement les dix-sept individus de Papouasie et les dix de l’Ile de Bougainville sont nettement plus proches que tous les autres non africains. Le bilan est donc que les Neandertaliens ont apporté 2,5 % de leur génome aux humains actuels hors d’Afrique et les Dénisoviens ont apporté 4,8 % de leur génome aux Papous ! Ce qui est difficile à expliquer, c’est pourquoi on ne retrouve pas d’ADN Dénisovien chez les autres populations d’Asie du Sud-est. Il faut sans doute faire l’hypothèse que les Dénisoviens peuplait une grande partie de l’Asie et qu’ils se sont métissés avec la première vague d’hommes modernes migrant le long des côtes vers l’Australie. Par ailleurs, la seconde vague de population moderne, métissée de Neandertal est venu peupler les autres territoires, pour au final apporer aux Papous un peu de Neandertal. Mais il est évident qu’un tel scénario doit être validé par d’autres recherches soit sur des hommes actuels, soit sur des fossiles venant de Chine ou d’autres contrées asiatiques. Récemment, une étude a montré que l’adaptation à l’altitude des populations tibétaines venait de la présence dans leur génome du gène EPAS1, dont ils ont hérité des Dénisoviens. Voila un nouveau venu dans la famille, l’homme de Denisova. C’est un cousin de l’homme de Neandertal, un peu plus âgé que lui. Et comme l’homme de Neandertal, il n’a pas complètement disparu puisqu’il continue à vivre dans l’ADN d’une partie au moins de la population actuelle. On le voit, le travail n’est pas terminé et des surprises peuvent encore se produire. Ce qui est exceptionnel dans cette dernière aventure, c’est qu’un morceau d’os, gros comme deux grains de riz, ait permis de découvrir une nouvelle humanité . Certes, la molaire découverte près de ce petit fragment du petit doigt d’une fillette mettait bien les archéologues sur la piste de l’homme de Denisova, mais c’est la connaissance de son génome qui a apporté la preuve de son existence. Le bouleversement du Néolithique Reste un dernier bouleversement dans l’histoire de l’avènement de l’humanité, c’est la révolution néolithique. La figure 4 exprime ce bouleversement par la croissance exponentielle de la population mondiale, passant en 10 000 ans d’environ 7 millions à 7 milliards d’hommes sur terre. Cette révolution est due à la maîtrise de l’élevage et de l’agriculture, permettant de libérer du temps, jusqu’alors essentiellement occupé à la chasse et à la cueillette. Alors ce temps ‘libre’ permettra la construction de sociétés, avec des préoccupations politiques, sociales, religieuses, … Les foyers de la néolithisation apparaissent sur la figure 15. C’est aux alentours de – 10 000 qu’en Turquie les premiers mouvements de cette révolution apparaissent, pour ce qui nous concerne, nous européens. Les techniques de l’élevage et de l’agriculture vont diffuser à travers l’Europe, en s’accompagnant d’un accroissement formidable de la population. La question est alors :
Il faut donc plutôt s’orienter vers la seconde hypothèse. En se basant sur des études de l’ADN-mt, il semblerait en fait qu’environ pour les ¾, la population européenne aurait une origine paléolithique C’est-à-dire qu’entre les deux hypothèses, la seconde serait prépondérante mais pas unique. Donc, les populations autochtones ont peu à peu adopté les techniques agricoles nouvelles, tout en étant peu métissées avec les populations qui leur apportaient ces nouvelles techniques. L’apport de ces nouvelles a permis le développement démographique dont nous avons déjà parlé. Mais ces études ont besoin d’être encore affinées, d’une part, et d’autre part d’être confrontées ce qui est connu par ailleurs, à savoir les courants de Néolithisation de l’Europe (voir figure 15). L’influence de la néolithisation sur les populations a-t-elle été la même suivant les deux courants ? Il semble que le métissage entre les populations autochtones et celles venant et apportant avec elles l’agriculture ait été plus marqué selon le courant méditerranéen que suivant le courant danubien. On peut penser par ailleurs que la Néolithisation des autres continents s’est faite de la même manière qu’en Europe, à savoir par diffusion (pacifique ?) des techniques plutôt que par diffusion des populations maîtrisant ces techniques innovantes. … et pour l’époque moderne Tous ces phénomènes de sélection ou de diffusion dans les populations, se traduisant dans une évolution spécifique du génome, ne se sont évidemment pas arrêtés avec l’époque ‘moderne’ (c’est-à-dire typiquement à l’ère chrétienne). Voici quelques exemples : Le nombre d’enfants par femme est évidemment limité. Par contre, des hommes peuvent être très prolifiques. De même les femmes (lors de mariages) changent plus facilement de groupe que les hommes. Ainsi, deux marqueurs très spécifiques du chromosome Y se trouve répartis dans une grande partie de l’Asie. L’origine de ces marqueurs date d’environ 1000 ans. Dans ce cas, la fréquence de ces marqueurs ne peut s’expliquer que par le fait que les porteurs de ces marqueurs avaient une descendance plus nombreuse que le reste des hommes dans ces populations. De plus les territoires, où ce phénomène est observé, correspond à l’extension de l’Empire de Gengis Khan ! On voit la solution : les hommes au pouvoir dans cet Empire avaient une descendance plus nombreuse que les autres hommes de l’Empire ! Mais ces deux marqueurs se retrouvent aussi au Pakistan (hors de l’Empire) dans une population … qui se dit descendante de Gengis Khan ! Autre exemple de sélection. Les Noirs américains sont plus sensibles à l’hypertension que leurs compatriotes, avec sensiblement le même régime alimentaire. Or, lors de la traite des Noirs, près des ¾ d’entre eux mouraient dans les premières années de leur transfert, en particulier lors de la traversée de l’Océan. Ceux qui résistaient le mieux étaient ceux qui avaient la capacité de mieux retenir le sel dans leur organisme, évitant ainsi la déshydratation. Ce qui fut un avantage est aujourd’hui un inconvénient, cette faculté les exposant maintenant aux maladies cardio-vasculaires. Cette sélection ‘artificielle’ fait qu’une population a vu son génome évoluer, positivement puis négativement. Ce dernier exemple illustre ce que l’on pourrait observer en étudiant les effets des grandes épidémies qui ont pu réduire des populations de manière catastrophique, les survivants pouvant avoir un génome particulier, significatif de leur résistance au fléau. On comprend alors l’intérêt que peut présenter la découverte de nécropoles du haut-Moyen-âge ou de l’époque romaine, avant certaines épidémies européennes. A condition que l’ADN ne soit pas dégradé ou contaminé par l’archéologue lui-même ! Ces exemples précédents exposent des effets permettant d’apporter des éclairages nouveaux à l’histoire. Mais il est d’autres utilisations qui se sont fait jour pour répondre à la curiosité des particuliers quant à l’origine de leurs ancêtres. Ainsi aux USA, des compagnies aériennes proposent aux Noirs américains de faire l’analyse de leur ADN pour leur faire savoir de quelle partie de l’Afrique était leur ancêtre (et on peut leur proposer le billet d’avion !). La méthode est plus performante pour indiquer quels sont ceux qui ont un ancêtre commun. En déduire une origine géographique est probabiliste, ce qu’acceptent mal certains clients. Toutefois, le nombre d’analyses se multipliant dans le monde, on peut penser que cette technique dans la recherche des origines particulières sera de plus en plus performante. Dans le même ordre d’idée, des canadiens se sont soumis à des recherches pour savoir si les porteurs d’un même nom avait un ancêtre immigrant commun. L’analyse de l’ADN-Y des homonymes fait penser qu’ils sont probablement issus d’un ancien immigrant arrivé 9 générations plus tôt. Par contre, l’ADN-mt, présente des caractères amérindiens ! Et ce n’est certainement pas fini ! Et l’histoire n’est peut-être pas aussi, simple ! Le peuplement de la terre est présenté avec des migrations hors d’Afrique. Mais s’est-il toujours fait dans le même sens, sans retour ? Une étude publiée en octobre 2015 vient encore bousculer ce qui est écrit jusqu’ici. Un squelette, vieux de 4 500 ans, a été découvert en Ethiopie. Il a été possible d’en extraire son ADN, celui d’un africain ancien. Or le génome de ‘Mota’ (ainsi l’a-t-on, appelé) est sensiblement différent de celui des africains actuels. Et les différences s’expliquent par un apport important (environ 7 %) venant des agriculteurs du Moyen-Orient. On savait que des migrations avaient eu lieu il y a environ 3 000 ans du Croissant fertile vers l’Afrique, bien après que ‘Mota’ soit décédé. Mais une telle influence sur le génome des africains traduit une ampleur insoupçonnée. Et ceci a une autre conséquence : ‘Mota’ est alors la référence africaine où la part du génome de Neandertal, qu’ont apporté avec eux ces agriculteurs venus de l’Eurasie, est nulle. Et on retrouve – en comparant le génome de ‘Mota’ à celui des africains actuels - des différences qui s’expliquent par un apport de Neandertal. Ce serait environ 0,2 à 0,7 % de l’ADN des africains qui aurait été emprunté indirectement aux Néandertaliens. Et ceci chez tous les africains. L’autre conséquence est celle-ci. Puisque le génome ‘moyen’ des africains d’aujourd’hui avait été pris comme référence pour un génome où l’apport du génome de Neandertal était nul, il faut augmenter d’autant chez les eurasiens d’aujourd’hui, de 0,2 à 0,7 %, l’apport du au métissage entre l’homme de Neandertal et l’homme moderne. Conclusion Depuis moins de dix ans, les progrès dans la connaissance de nos ancêtres ont été fulgurants. Cette nouvelle science apporte indiscutablement un éclairage nouveau. L’histoire de l’homme est sans doute complexe. Mais ce que l’on a aussi découvert, c’est que tous ces ancêtres que l’on disait disparus, tels quel l‘homo erectus ou l’homme de Neandertal, vivait encore en nous, dans notre ADN. Ils n’ont donc pas complètement disparus. Il y a un avant Avant d’être nés à nous-mêmes, nous sommes nés des autres et nés aux autres. Et les autres font partie de nous. Nous sommes faits de l’empreinte de ce qui a disparu, de celles et de ceux qui ont disparu. Nous sommes faits de la présence de l’absence, de ce qui demeure en nous de tous ceux qui nous ont précédés. Nous sommes des héritiers. Mais des pans entiers de cet héritage se sont effacés. Aller à la rencontre de notre passé. Aller à notre propre rencontre. Jean-Claude Ameisen Bibliographie : (L’auteur doit beaucoup aux ouvrages suivants) ‘Anthropobiologie, Évolution humaine’, E. Crubézy, J. Braga et G. Larrouy. Elsevier-Masson, 2008. ‘Quand d’autres hommes peuplaient la terre. Nouveaux regards sur nos origines’. J.-J. Hublin, Flammarion, 2011 ‘Neandertal, A la recherche des génomes perdus’, Svante Pääbo, Les Liens qui libèrent’2015. ‘Sur les Epaules de Darwin – Retrouver l’aube’, Jean-Claude Amaisen, Les Liens qui libèrent’2014. ‘Madame de Neandertal – Journal intime’, Pascale Leroy et Marylène Pathou-Mathis’, NiL Editions, 2014. |
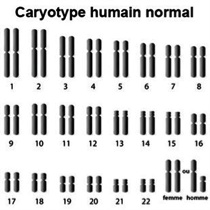
1 : Description du caryotype humain.
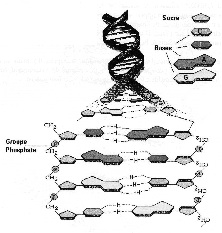
2 : La molécule d’ADN (vue partielle !)
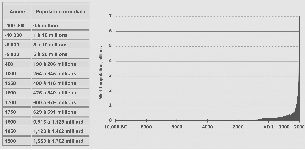
3 – Tableau de l’évolution de la population mondiale au cours du temps.
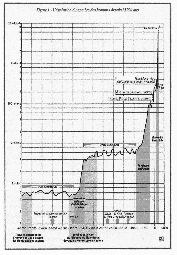
4 : La population mondiale en coordonnées log-log (d’après l’INED) °
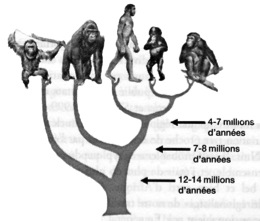
5 : La superfamille des Hominoïdes
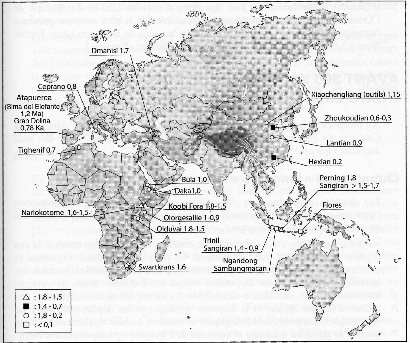
6 : Répartition et dates des fossiles d’homo erectus dans le monde.
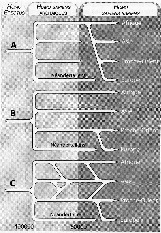
7 : Hypothèses sur l’origine de l’homme moderne

8 : distribution géographique des principaux haplogroupes de l’ADN-Y

9 : Arbre phylogénétique des haplogroupes de l’ADN-Y
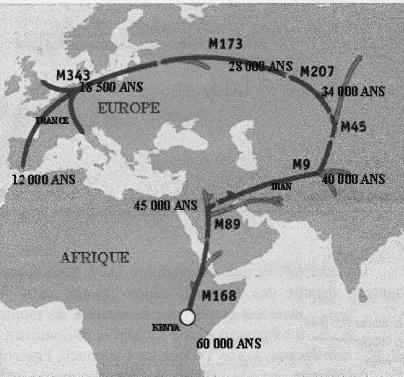
10 : Parcours d’homo sapiens sapiens vers l’Europe
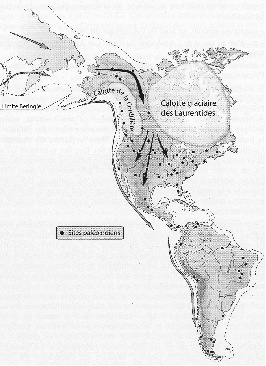
11 : Schéma de colonisation de l’Amérique.

12 : Arbre phylogénétique construit à partir des premiers résultats obtenus sur l’ADN-mt de l’homme de Neandertal. L’ « Eve mitochondriale » est positionnée par le point gris. Les chiffres indiquent le critère statistique.
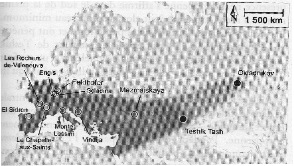
13 : Zone d’expansion de Neandertal, avec mention des sites où des fossiles ont permis l’étude de l’ADN.
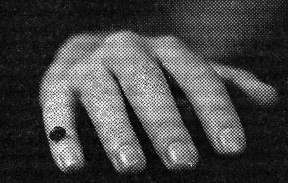
14 : Fragment d’os de Denisova, un morceau de la dernière phalange de l’auriculaire
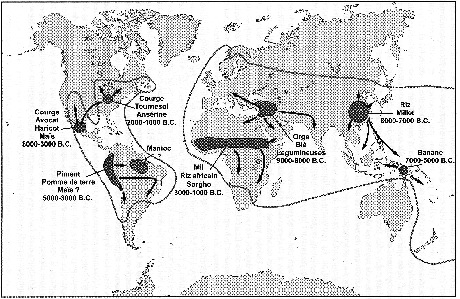
15 : Foyers de Néolithisation à travers le monde et diffusion.
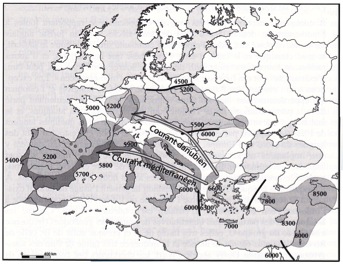
16 : Les courants de Néolithisation en Europe
|